 Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021)
Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021) Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)
Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)Regulation of Stroke Volume
Ventricular stroke volume (SV) is often thought of as the amount of blood (mL) ejected per beat by the left ventricle into the aorta (or from the right ventricle into the pulmonary artery). This assumes, however, that all the blood leaving the ventricle is ejected into the outflow tract, but this is not the case when there is atrioventricular valve regurgitation or an interventricular septal defect. Therefore, a more precise definition for SV and one that is used in echocardiography when assessing ventricular function is the difference between the ventricular end-diastolic volume (EDV) and the end-systolic volume (ESV). The EDV is the filled volume of the ventricle before contraction, and the ESV is the residual volume of blood remaining in the ventricle after ejection. In a typical heart, the EDV is about 120 mL of blood and the ESV is about 50 mL of blood. The difference in these two volumes, 70 mL, represents the SV (SV = EDV – ESV). Therefore, any factor that alters either the EDV or the ESV will change the SV. For example, an increase in EDV increases SV, whereas an increase in ESV decreases SV.
There are three primary mechanisms that regulate EDV and ESV, and therefore SV. These are preload, afterload, and inotropy.
Preload
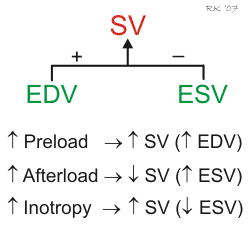 Changes in preload affect the SV through the Frank-Starling mechanism. Briefly, an increase in venous return to the heart increases the filled volume (EDV) of the ventricle, which stretches the muscle fibers, increasing their preload. This leads to an increase in the force of ventricular contraction and enables the heart to eject the additional blood that was returned to it. Therefore, an increase in EDV results in an increase in SV. Conversely, a decrease in venous return and EDV leads to a decrease in SV by this mechanism.
Changes in preload affect the SV through the Frank-Starling mechanism. Briefly, an increase in venous return to the heart increases the filled volume (EDV) of the ventricle, which stretches the muscle fibers, increasing their preload. This leads to an increase in the force of ventricular contraction and enables the heart to eject the additional blood that was returned to it. Therefore, an increase in EDV results in an increase in SV. Conversely, a decrease in venous return and EDV leads to a decrease in SV by this mechanism.
Afterload
Afterload is related to the pressure that the ventricle must generate to eject blood into the aorta. Changes in afterload alter the ability of the ventricle to eject blood and alter ESV and SV. For example, an increase in afterload (e.g., increased aortic pressure) decreases SV, and causes ESV to increase. Conversely, a decrease in afterload augments SV and decreases ESV. It is important to note, however, that the SV in a normal, non-diseased ventricle is not strongly influenced by afterload because of compensatory changes in preload. In contrast, the SV of hearts that are failing are very sensitive to changes in afterload.
Inotropy
Changes in ventricular inotropy (contractility) alter the rate of ventricular pressure development, and ESV and SV. For example, an increase in inotropy (e.g., produced by sympathetic activation of the heart) increases SV and decreases ESV. Conversely, a decrease in inotropy (e.g., heart failure) reduces SV and increases ESV.
It is important to note that the effects of EDV and ESV on SV are interdependent. For example, an increase in ESV usually results in a compensatory increase in EDV. If SV is increased by increasing EDV, this can lead to a small increase in ESV because of the influence of increased afterload on ESV caused by an increase in aortic pressure. Therefore, while the primary effect of a change in preload, afterload or inotropy may be on either EDV or ESV, secondary changes can occur that can partially compensate for the initial change in SV. For a more detailed description of these interactions, see the pages describing preload, afterload, or inotropy.
Revised 02/05/2024